-
The Machinery Within
Inside every cell, life depends on assemblies of proteins coming together like small, deliberate machines. They twist, fold, lock, release — a natural machinery hidden in plain sight. We are building AI models to reveal them.

-
Viral Spike Shapes
Viral spike proteins must remain stable while outside cells, but undergo massive transitions to trigger cell entry once they encounter a host receptor.
-

Oncogenic Signaling Systems
Cancer is not just a structural defect in one molecule — it is a failure of a distributed control system that normally maintains balance.
-

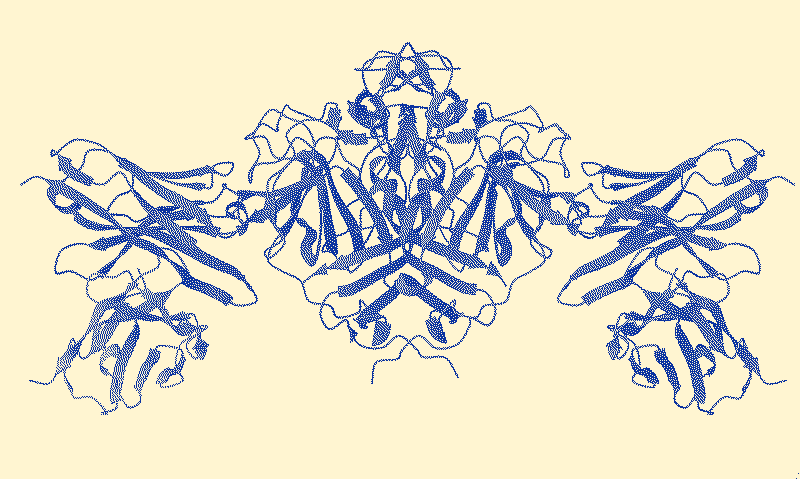
Making Protein Surfaces Legible
Fab fragments act as molecular stabilizing grips, allowing researchers to turn unstable protein behaviors into readable structures.
-
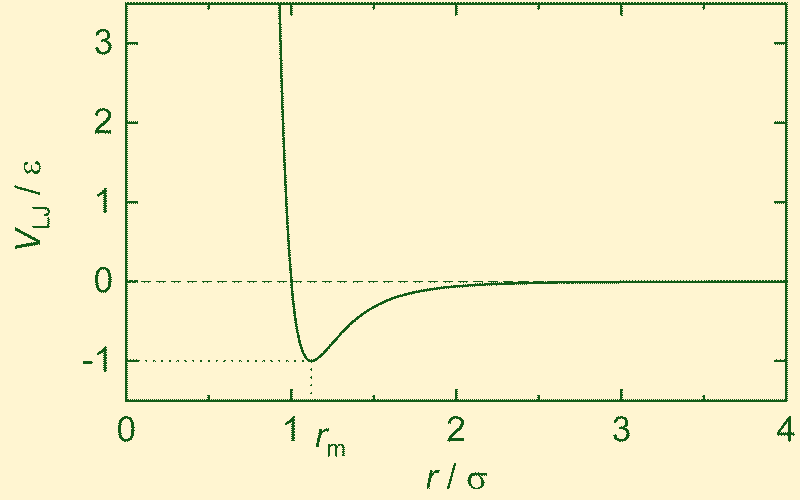
The Lennard–Jones Potential
A fundamental tension present throughout nature: entities repel when forced too close together, yet attract one another at intermediate distances.
-
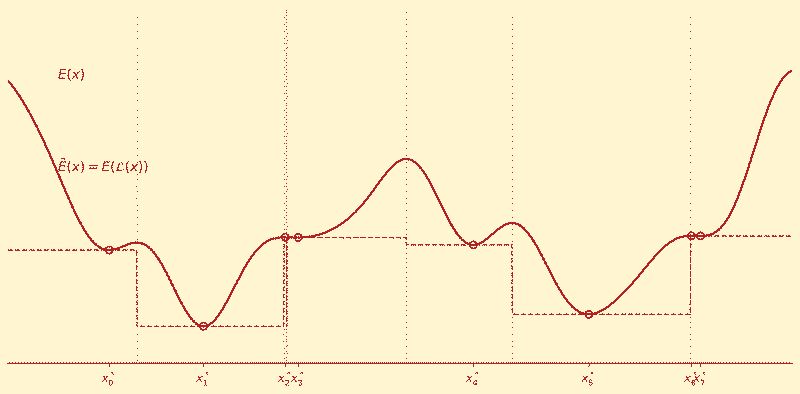
Monte Carlo Basin Hopping
Instead of trying to descend a complex landscape directly, repeatedly perturb the system and re-minimize locally.
-

A Profound Shift
AlphaFold changed the timeline of structural biology overnight. What felt 200 years away began to take shape in front of us.
-

Our Work
Predicting quaternary structure — how multiple protein subunits come together to form functional complexes — is the next frontier.
-

Signed Distance Function (SDF)
Rather than describing an object by its surface, vertices, or volume, it describes every point in space by a single quantity: distance.
San Francisco, CA
Jäntra Biosystems, Inc.
Jäntra Biosystems, Inc.
 jäntra.ai
jäntra.ai
Revealing the Machinery of Life